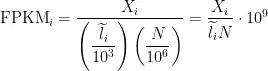I have seen many posts regarding counts to RPKM and TPM. I haven't seen any post for counts to FPKM.
I have RNA-Seq data which is paired-end reads. Extracted the counts using featureCounts for all the samples.
There is a function to convert counts to RPKM: using the gene_length
rpkm <- function(counts, lengths) {
rate <- counts / lengths
rate / sum(counts) * 1e6
}
I know that RPKM is mainly used for single-end reads data. Do you think I can use the above function for converting counts to FPKM as my data is paired end? [TCGA data: HTSeq counts are converted to HTSeq FPKM]
Somewhere I have seen that the same function applies for single end data which will be RPKM and also for paired end data which will be FPKM. Am Ir right or wrong?
If not can anyone show some function or code to convert counts to FPKM please.
thanq